Cardiomiopatia hipertrófica: tudo que você precisa saber

Você já deve ter ouvido falar sobre a cardiomiopatia hipertrófica. Em uma primeira impressão, a doença pode assustar pela complexidade, mas vamos te mostrar tudo sobre ela para te preparar para identificá-la e tratá-la nos pacientes. Confira a seguir!
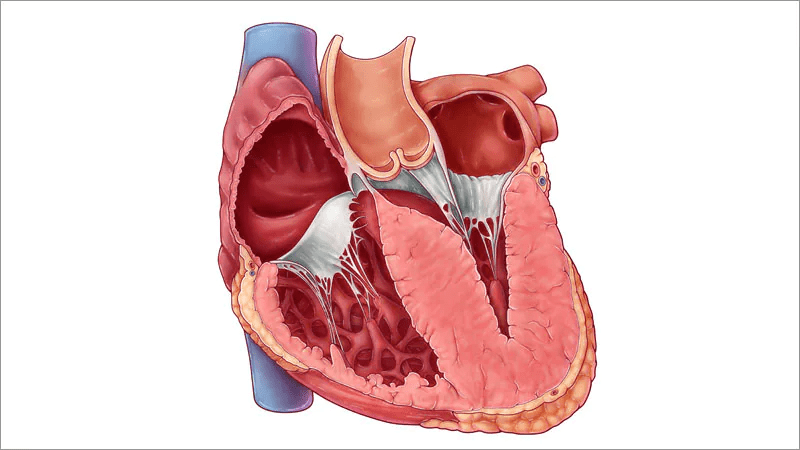
Definição de cardiomiopatia hipertrófica
A cardiomiopatia hipertrófica (CMH) é uma doença cardíaca primária, genética, autossômica dominante, de expressão fenotípica variável. Ela é caracterizada pela presença de hipertrofia ventricular esquerda concêntrica na ausência de qualquer outra doença cardíaca ou sistêmica que justifique o grau de hipertrofia.
Ela possui as seguintes características:
- hipertrofia miocárdica assimétrica (principalmente da parede ântero-lateral e septal);
- dilatação e hipertrofia do átrio esquerdo;
- fibrose intersticial;
- alteração na vasculatura.
É uma das doenças genéticas mais comuns, afetando 0,2% da população geral e 0,5% dos portadores de cardiopatia. Além disso, é mais vista em brancos e do gênero masculino.
Podemos afirmar que a CMH é transmitida de forma genética autossômica dominante em até 60% das vezes. Os demais casos são formas esporádicas de etiologia pouco esclarecida.
São mais de 400 mutações identificadas, sendo que as principais são: gene da cadeia pesada da beta miosina 7 cardíaca (35-55% dos casos), gene da proteína C3 de ligação à miosina ( 20-25%) e gene da troponina T cardíaca (15% dos casos).
Fisiopatologia
A fisiopatologia não é totalmente esclarecida, mas supõe-se que ocorram eventos moleculares desencadeados por mutações genéticas. Sendo assim, foram levantadas duas teorias:
- o gene defeituoso interfere na função do gene normal;
- o gene defeituoso adquire propriedades de dominar a função do gene normal, gerando como resultado uma insuficiência na produção de proteínas normais.
As proteínas anômalas encontram-se em ambos ventrículos, porém em maior quantidade no VE, câmara de maior pressão. Por diversos mecanismos, favorece a atuação de fatores determinantes de crescimento miocárdico, como peptídeo natriurético atrial e cerebral, endotelina e sistema renina-angiotensina-aldosterona.
Em resposta à atuação desses fatores, ocorre hipertrofia dos miócitos, proliferação de fibroblastos e consequente acúmulo de colágeno, além de alterações na microcirculação. Tudo isso resulta em um ventrículo rígido, com comprometimento da função sistólica e diastólica, apesar de uma fração de ejeção inicialmente preservada do VE.
Além disso, ocorrem outras alterações: obstrução da via de saída dinâmica do VE, regurgitação mitral, disfunção diastólica, isquemia miocárdica, arritmias e disfunção autonômica. A seguir, conheça cada uma delas!
Obstrução da via de saída do VE (OVSVE)
Está presente em cerca de 75% dos casos e envolve 2 mecanismos: hipertrofia septal com estreitamento da via de saída e alterações anatômicas do aparelho mitral.
Tais mecanismos ocasionam movimentação anormal do folheto mitral durante a sístole, gerando regurgitação e aumento da pressão intracavitária, que estimula a hipertrofia da musculatura e causa isquemia miocárdica.
A carga ventricular e a contratilidade influenciam o gradiente pressórico, sendo variáveis diretamente proporcionais. A presença de um pico de gradiente da VSVE maior ou igual a 30 indica obstrução.
Disfunção diastólica
A alta pressão intracavitária, a contratilidade e o relaxamento irregular, associados a uma recaptação anormal de cálcio intracelular, ocasionam a disfunção diastólica. As alterações são secundárias a fibrose e mudanças anatômicas.
Regurgitação mitral
É secundária à OVSVE ou às anormalidades do folheto primário e contribui para o surgimento de sintomas, principalmente, a dispneia. A direção do jato da regurgitação define a forma do tratamento nos casos cirúrgicos.
Isquemia miocárdica
Ocorre em decorrência do desbalanço entre a oferta e a demanda de oxigênio. Os principais fatores que geram esse desequilíbrio são a hipertrofia do miocárdio, a disfunção da microvasculatura com reserva de fluxo coronariano comprometida e a hipertrofia medial das arteríolas intramurais.
A isquemia miocárdica predispõe a formação de aneurismas do VE. Em decorrência disso, há o aumento do risco de arritmias e insuficiência cardíaca (IC).
Disfunção autonômica
Está relacionada a uma resposta inadequada da pressão arterial, da frequência cardíaca e da vasodilatação arterial ao esforço físico, fatores que podem levar a um pior prognóstico.
Quadro clínico
Agora que você viu a etiologia e a fisiopatologia, vamos detalhar os sintomas de cardiomiopatia hipertrófica. A suspeita inicia-se quando há a presença de histórico familiar (em pelo menos três gerações), sinais e sintomas de IC, que surgem durante esforço físico, evento cardíaco coronariano e alterações em exames complementares, como ECG.
Contudo, é assintomática na maioria dos pacientes. Por meio do exame físico é possível identificar pistas para prosseguir a investigação. Fique atento à presença de sopro de insuficiência mitral, pulso carotídeo em martelo d’água e quarta bulha.
Lembrando que você pode lançar mão de manobras provocativas, como a de Valsalva, por exemplo, para melhor avaliação. O eletrocardiograma pode dar dicas também. A partir dele, há chances de identificar sinais de hipertrofia do VE, distúrbios de repolarização e arritmias. Contudo, as alterações não se correlacionam de forma confiável com a gravidade do quadro.
Por outro lado, ele pode ser usado como teste de triagem em pessoas com histórico familiar, uma vez que tem a capacidade de mostrar alterações que antecedem a hipertrofia do VE.
A presença de arritmias, principalmente a fibrilação atrial e a taquicardia ventricular não sustentada, indica pior prognóstico e determina mudança tanto no seguimento quanto no tratamento dos pacientes. Após avaliação clínica, pode ser indicada a realização do holter.
Diagnóstico
O diagnóstico deve ser feito por meio de exames de imagem. O ecocardiograma e a ressonância magnética cardíaca mostram uma espessura da parede diastólica final máxima maior ou igual a 15 mm em qualquer lugar do VE, na ausência de outra causa de hipertrofia em adultos.
Em crianças, deve-se ajustar pelo tamanho e pelo crescimento corporal, considerando um escore Z maior ou igual a dois, na presença de história familiar, ou teste genético positivo (na ausência desses sinais, considerar escore maior ou igual a 2,5).
O padrão de distribuição de espessamento é variável, podendo ser limitado e focal. O local mais comum é o septo anterior basal em continuidade com a parede anterior livre.
Podem ocorrer outras alterações associadas, como a hipertrofia, o deslocamento apical dos músculos papilares, as criptas miocárdicas, a ponte miocárdica, a hipertrofia do VD e os folhetos alongados da valva mitral.
Exames para o diagnóstico
O ecocardiograma identifica as alterações fisiopatológicas. Entre elas, estão o grau da obstrução da via de saída do VE, as disfunções sistólicas e diastólicas, as possíveis áreas de isquemia e complicações (como o aneurisma do VE, por exemplo), a função da valva mitral e a estima da fração de ejeção.
A RMN cardíaca ajuda a identificar as diversas expressões fenotípicas da CMH, realiza predição de risco, interfere no planejamento pré-cirúrgico e avalia alterações estruturais cardíacas.
Além disso, pode detectar a disfunção diastólica pelo realce tardio com gadolínio (que representa a fibrose miocárdica), que também pode sugerir áreas de isquemia miocárdica. É um importante exame quando a história é sugestiva, mas o ecocardiograma é normal ou inconclusivo.
Exames complementares
A tomografia cardíaca é outro exame que pode ser solicitado quando a RNM cardíaca se encontra indisponível ou contraindicada. Avaliar a estrutura cardíaca e a perfusão coronariana, porém tem menor sensibilidade quando comparada a RNM.
A cineangiocoronariografia é um exame de exceção na investigação da CMH. É indicada quando os testes não invasivos se mostram ineficazes ou com baixa acurácia para apontar alterações cardíacas que poderiam modificar o tratamento. Ainda é realizada ao optar por tratamento cirúrgico em pacientes portadores de diversos fatores de risco para DAC.
Triagem genética
A triagem genética deve ser realizada em todo paciente que possui história familiar ou eventos suspeitos de CMH durante investigação de pelo menos três gerações. Ela é importante na confirmação diagnóstica e interfere no aconselhamento genético dos pacientes portadores.
Alguns pacientes podem apresentar genótipo positivo e fenótipo negativo, representando o estágio pré-clínico da CMH e relacionando-se com um seguimento mais rigoroso.
Teste de esforço
Quando associado à análise simultânea dos gases respiratórios (teste cardiopulmonar de exercício), o teste de esforço traz informações quanto à gravidade e ao mecanismo de limitação funcional. Ele é importante em pacientes com indicação de transplante cardíaco e assintomáticos, para avaliar se há obstrução da via de saída do VE.
Tratamento farmacológico
O tratamento da cardiomiopatia hipertrófica pode ser feito de duas maneiras. A primeira delas é pelo uso de fármacos indicados após o diagnóstico e a realização de exames. Entre eles, estão:
- beta bloqueadores na dose máxima tolerada em pacientes com CMH obstrutiva e sintomática. Na ausência de resposta, substituir por bloqueador de canal de cálcio não dihidropiridínico. Também está indicado nos casos de angina associada a uma FE preservada. Não está bem estabelecido o uso em pacientes assintomáticos;
- uso cauteloso de diuréticos em pacientes com CMH obstrutiva e dispneia persistente ou presença de sintomas congestivos;
- agentes vasoconstritores em casos de hipotensão aguda, sendo uma urgência médica;
- na presença de ICFER, guiar terapia com base na IC e tratar causas concomitantes (se presentes);
- na presença de FA, deve ser realizada anticoagulação, sendo a primeira linha os novos anticoagulantes orais. Em casos que for optado por controle da frequência, priorizar os beta bloqueadores. O controle do ritmo, por medicamentos, cardioversão ou ablação, deve ser discutido em conjunto com o paciente, ponderando os riscos e os benefícios.
Além disso, agentes vasodilatadores podem causar piora dos sintomas obstrutivos, portanto, deve-se considerar a suspensão dessas medicações. O uso do IECA não tem benefício bem estabelecido;
Tratamento invasivo
Outra forma de tratamento da cardiomiopatia hipertrófica é o método invasivo. Ele consiste na terapia de redução septal e está indicado nos seguintes casos:
- pacientes sintomáticos refratários a terapia clínica;
- outra doença cardíaca com necessidade de tratamento cirúrgico (p.ex.: DAC multiarterial);
- hipertensão pulmonar grave e progressiva secundária à OVSVE;
- aumento do átrio esquerdo com um ou mais episódios de FA sintomática;
- baixa capacidade funcional comprovada em teste de esforço físico;
- gradiente de repouso da VSVE acima de 100 mmHg em crianças e adultos jovens;
- CDI indicado nos casos de TVNS, morte súbita abortada, morte súbita secundária à CMH em um ou mais parentes de primeiro grau ou próximos com idade inferior a 50 anos, HVE maior ou igual a 3 cm em qualquer segmento do VE, pelo menos um episódio de síncope relacionado a presença de arritmia, aneurisma apical do VE e disfunção sistólica do VE associada a uma fração de ejeção reduzida.
Pacientes diagnosticados devem realizar seguimento conforme a presença ou não dos sintomas. Em pacientes assintomáticos, deve-se repetir o ecocardiograma a cada um ou dois anos, não existindo consenso a respeito do tempo de seguimento.
É importante realizar em pacientes com história familiar sugestiva e teste genético positivo, a despeito de estarem assintomáticos. Nos pacientes sintomáticos, o seguimento deve ser individualizado.
O prognóstico da CMH depende da presença de eventos adversos, tais como insuficiência cardíaca com sintomas limitantes, AVE, arritmia ventricular ou fibrilação atrial. A presença de um ou mais eventos adversos aumenta a mortalidade. Contudo, com o tratamento e o rastreio precoce dos marcadores de risco, a mortalidade cai e chega a ser menor que 1% ao ano.
Amplie seu conhecimento com a gente!
Gostou de saber mais sobre a cardiomiopatia hipertrófica? Então, aproveite para conferir mais conteúdos como este na Academia Medway. Aqui, você encontra e-books e minicursos completamente gratuitos.
Se quiser acumular mais conhecimento ainda sobre a área, o PSMedway, pode ser uma boa opção. Em nosso curso, vamos te mostrar exatamente como é a atuação médica na sala de emergência. Então, não perca tempo!


Hellen Sanches Pitaluga
Meu nome é Hellen Sanches Pitaluga e sou natural de Goiânia. Sou formada pela Universidade de Ribeirão Preto desde 2019. Residência em Clínica Médica no Complexo Hospitalar Prefeito Edivaldo Orsi da Rede Mário Gatti em Campinas, São Paulo. Amo clínica médica e sua diversidade, mas quando se trata da cardiologia o há um pouco mais de amor.
Você também pode gostar




