Metabolismo do Etanol: rota bioquímica e as implicações clínicas da intoxicação por álcool

Você já parou para pensar no que acontece com o álcool depois do primeiro gole? Embora o consumo da substância esteja profundamente inserido em diferentes contextos sociais e culturais, o metabolismo do etanol envolve uma rede complexa de reações bioquímicas com efeitos que vão muito além do fígado.
Ao ser absorvido e distribuído pelo organismo, o etanol interage com enzimas, altera vias metabólicas, modifica o equilíbrio celular e repercute diretamente no funcionamento do sistema nervoso.
Entender esse processo é essencial para compreender, de forma integrada, como uma substância tão comum pode produzir efeitos tão amplos no corpo humano.
Em relação à epidemiologia, o consumo de álcool segue altamente prevalente e clinicamente relevante.
Um estudo brasileiro de série temporal mostrou aumento do consumo abusivo de álcool nas capitais do país, de 15,6% em 2006 para 20,8% em 2023, o que representa aumento relativo de cerca de 33% (Malta et al., 2024). O crescimento foi especialmente marcante entre mulheres, com tendência de alta observada em 23 capitais brasileiras.
No cenário global, a mensagem é semelhante. Em análise internacional publicada em 2025, o consumo médio de álcool entre adultos passou de 5,1 litros per capita em 2000 para 5,5 litros em 2019, um aumento de aproximadamente 7,8% (Shield et al., 2025). No mesmo estudo, o álcool esteve associado a 2,6 milhões de mortes em 2019, correspondendo a 4,7% de todas as mortes, além de 116 milhões de DALYs perdidos.
Assim, além de sua relevância acadêmica, o etanol permanece como importante problema de saúde pública (Shield et al., 2025).
Onde o etanol é metabolizado e por que isso importa?
A maior parte do metabolismo do etanol ocorre no fígado. A principal via começa com a conversão do etanol em acetaldeído pela enzima álcool desidrogenase, a famosa ADH, localizada no citosol do hepatócito.
Depois, o acetaldeído é oxidado a acetato pela aldeído desidrogenase, sobretudo a ALDH2 mitocondrial. Em paralelo, existe a via do sistema microssomal oxidante do etanol, mediada principalmente pelo P4502E1 ou CYP2E1 presentes nas células hepáticas, que ganha relevância em concentrações mais altas de álcool e no uso crônico. A catalase participa de forma menor.
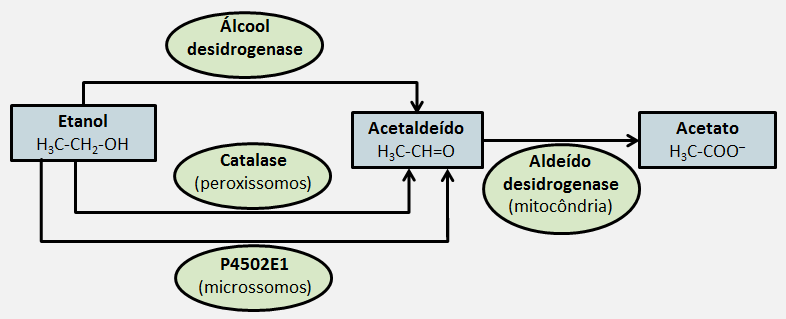
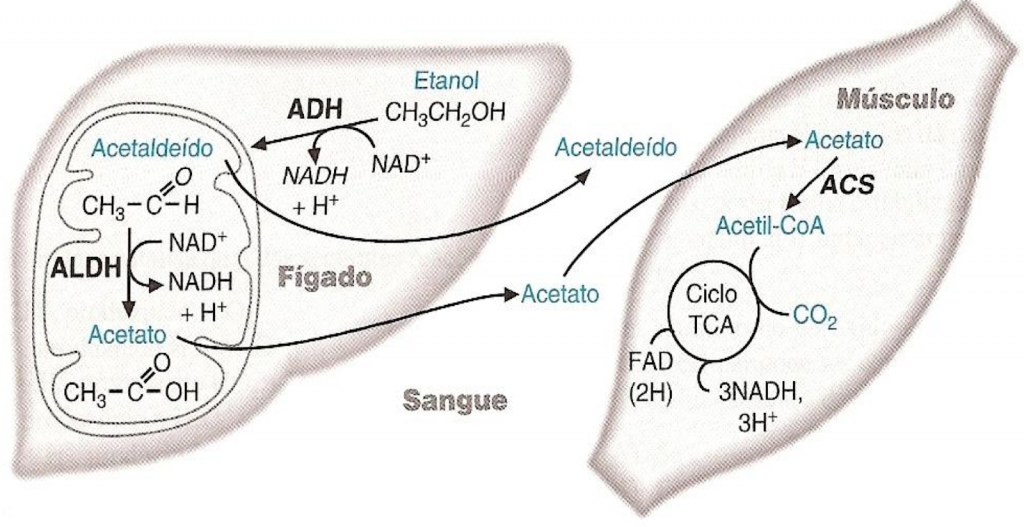
Até aqui, tudo parece uma sequência enzimática simples. O problema é o que essa sequência faz com o organismo. Depois de ser convertido em acetaldeído e, posteriormente, em acetato, esse metabólito passa a ser utilizado por diferentes tecidos, como coração e músculo esquelético.
Nesses locais, a acetil-CoA sintetase promove sua ativação em acetil-CoA, na matriz mitocondrial, permitindo que ele seja direcionado para múltiplas vias, incluindo o ciclo de Krebs: síntese de ácidos graxos, formação de corpos cetônicos e síntese de colesterol. Ao mesmo tempo, a oxidação do etanol e do acetaldeído leva à produção excessiva de NADH, com aumento da razão NADH/NAD+.
O resultado é um importante desbalanço redox celular, ou seja, uma alteração no equilíbrio entre compostos oxidados e reduzidos dentro da célula.
Esse fenômeno é central para a fisiopatologia do etanol, pois interfere diretamente em vias metabólicas essenciais e contribui para manifestações como hipoglicemia, acidose láctica e acúmulo de gordura no fígado, como veremos.
O que o excesso de NADH provoca na prática?
Quando o NADH aumenta, o piruvato tende a ser convertido em lactato.
Resultado: maior risco de acidose láctica. Ao mesmo tempo, a gliconeogênese fica prejudicada, porque faltam intermediários disponíveis para manter a produção hepática de glicose.
É por isso que o paciente que bebeu muito, ficou em jejum, vomitou e chega confuso ao pronto-socorro pode evoluir com hipoglicemia.
O mesmo raciocínio vale para o metabolismo lipídico. O excesso de NADH inibe a beta-oxidação de ácidos graxos e favorece acúmulo de triglicerídeos no fígado, contribuindo para esteatose hepática.
Outro desfecho clássico é a cetoacidose alcoólica, que pode ocorrer após ingestão importante de álcool associada a jejum e vômitos persistentes. Nesse contexto, a depleção de glicogênio, o aumento de hormônios contrarreguladores e a alteração redox favorecem a cetogênese, com elevação de beta-hidroxibutirato.
Clinicamente, o quadro pode cursar com dor abdominal, náuseas, taquipneia e acidose metabólica com ânion gap elevado, geralmente com glicemia normal ou discretamente baixa.
Como o álcool age no cérebro?
No sistema nervoso central, o etanol não funciona apenas como “depressor inespecífico”. Ele interage com sistemas de neurotransmissão muito bem definidos. Em termos simplificados, o álcool potencializa a neurotransmissão GABAérgica e reduz a atividade glutamatérgica, especialmente via receptores NMDA.
Essa combinação aumenta a inibição neural e explica os efeitos agudos típicos da intoxicação: sedação, lentificação psicomotora, fala pastosa, ataxia, prejuízo de atenção e redução de reflexos.
Além disso, o etanol modula circuitos de recompensa envolvendo dopamina, peptídeos opioides endógenos e serotonina. Esse efeito ajuda a entender por que o álcool produz sensação inicial de relaxamento, desinibição e reforço positivo.
Na prática, isso se conecta diretamente à base neurobiológica do transtorno por uso de álcool.
Por que a abstinência alcoólica pode ser tão grave?
Aqui entra um conceito muito interessante: neuroadaptação. Com o uso crônico, o cérebro tenta compensar a ação depressora do álcool. Há redução relativa da resposta inibitória e aumento compensatório da excitabilidade glutamatérgica.
Quando o etanol é retirado de forma abrupta, o sistema nervoso central fica “sem freio”, em estado hiperexcitatório. É daí que surgem tremores, ansiedade, insônia, sudorese, taquicardia, convulsões e, mais grave, o delirium tremens.
O cérebro também metaboliza etanol?
Sim, e esse ponto tem ganhado destaque em revisões recentes. Embora o fígado seja o principal local de metabolização, o cérebro também participa desse processo, com papel relevante de astrócitos, mitocôndrias e sistemas oxidativos locais.
Isso contribui para geração de acetaldeído, estresse oxidativo, disfunção mitocondrial e neuroinflamação. Em outras palavras, o dano neurológico do álcool não é apenas secundário à doença hepática ou à carência nutricional: existe neurotoxicidade direta.
Com exposição crônica, isso pode se traduzir em déficit cognitivo, comprometimento de função executiva, alterações de memória e dano estrutural cerebral.
É por isso que o tema também dialoga com neurologia e psiquiatria, e não apenas com hepatologia.
Variabilidade genética: por que algumas pessoas “passam mal” mais rápido?
Atenção, nem todo mundo metaboliza etanol da mesma forma. Polimorfismos em genes como ADH1B e ALDH2 alteram a velocidade de produção e depuração do acetaldeído.
A variante ALDH2*2, bastante prevalente em populações do Leste Asiático, reduz a capacidade de detoxificação do acetaldeído. O resultado é o quadro clássico de flushing facial, taquicardia, náuseas e mal-estar após ingestão alcoólica.
Esse detalhe é ouro para integrar genética e clínica. O acúmulo de acetaldeído torna a experiência de beber mais aversiva, o que pode reduzir risco de consumo continuado em alguns indivíduos.
Por outro lado, se o consumo persiste, a maior exposição ao acetaldeído aumenta o potencial de dano celular e mutagênese. O acetaldeído, afinal, é uma molécula tóxica e pró-carcinogênica, capaz de induzir lesão ao DNA.
E no neurodesenvolvimento?
A exposição pré-natal ao álcool é uma das faces mais graves desse tema. O etanol interfere em proliferação celular, migração neuronal, diferenciação, sinaptogênese e plasticidade cerebral.
As consequências podem se manifestar ao longo do espectro dos transtornos fetais relacionados ao álcool, com prejuízos cognitivos, comportamentais e adaptativos persistentes.
Do ponto de vista prático, a mensagem para a formação médica é objetiva: não há nível comprovadamente seguro de consumo alcoólico na gestação.
Esse é um conceito importante tanto para aconselhamento clínico quanto para provas que cobram prevenção e saúde da mulher.
Principais complicações clínicas para lembrar
Na intoxicação aguda, os principais riscos são rebaixamento do nível de consciência, trauma, broncoaspiração, depressão respiratória, hipoglicemia e distúrbios ácido-base.
Já no uso crônico, entram em cena esteatose, hepatite alcoólica, cirrose, pancreatite, cardiomiopatia, neuropatia periférica, transtornos psiquiátricos e deficiência de tiamina.
Não podemos esquecer do delirium tremens, que é o quadro mais grave da abstinência alcoólica, caracterizado por confusão mental aguda, agitação e hiperatividade autonômica após suspensão abrupta do consumo crônico de álcool.
A deficiência de tiamina merece menção especial. Pacientes com uso crônico de álcool, desnutrição e alteração neurológica, é obrigatório pensar na encefalopatia de Wernicke, especialmente diante de ataxia, oftalmoparesia, nistagmo e confusão mental.
Na prática, isso tem implicação direta na conduta: a tiamina deve ser administrada precocemente, idealmente antes ou junto com a glicose, para reduzir o risco de piora metabólica cerebral.
Como esse tema cai na prova?
Se você estiver no ciclo básico, a prova costuma cobrar as enzimas do metabolismo do etanol, o papel do NADH e as repercussões bioquímicas.
Já nas provas de residência, é muito comum a integração com cenários clínicos: paciente em jejum que faz hipoglicemia após etilismo, quadro de abstinência alcoólica, cetoacidose alcoólica, deficiência de tiamina e lesão hepática.
O metabolismo do etanol é um daqueles assuntos em que a bioquímica deixa de ser abstrata e vira raciocínio. A via ADH–ALDH não explica apenas como o álcool é eliminado, mas por que ele gera acetaldeído tóxico, altera o estado redox celular, favorece hipoglicemia e acidose, modula neurotransmissores e causa neuroadaptação.
Quando você entende essa lógica, fica mais fácil interpretar desde um paciente intoxicado na emergência até um caso de abstinência, hepatopatia alcoólica ou exposição fetal ao álcool.


Beatriz Stephan Farhat Jorge
Graduada em Medicina pela Universidade Federal de Juiz de Fora (UFJF), com residência médica em Clínica Médica pelo Hospital Albert Einstein.


